References:
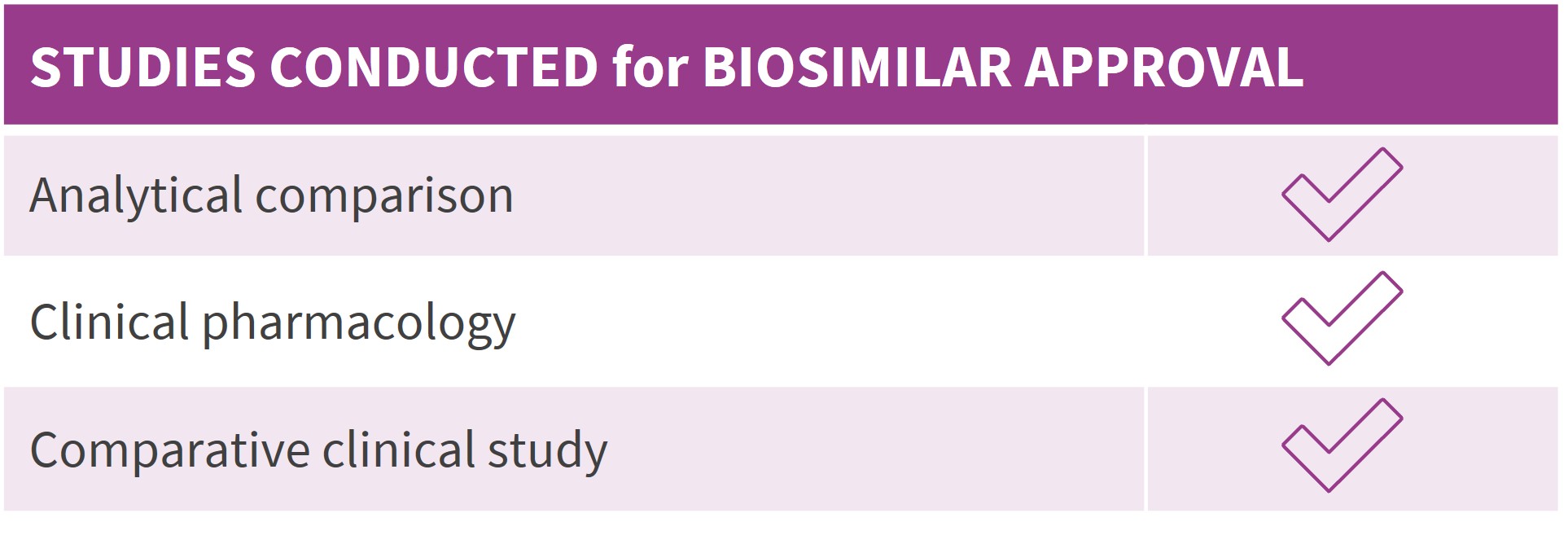
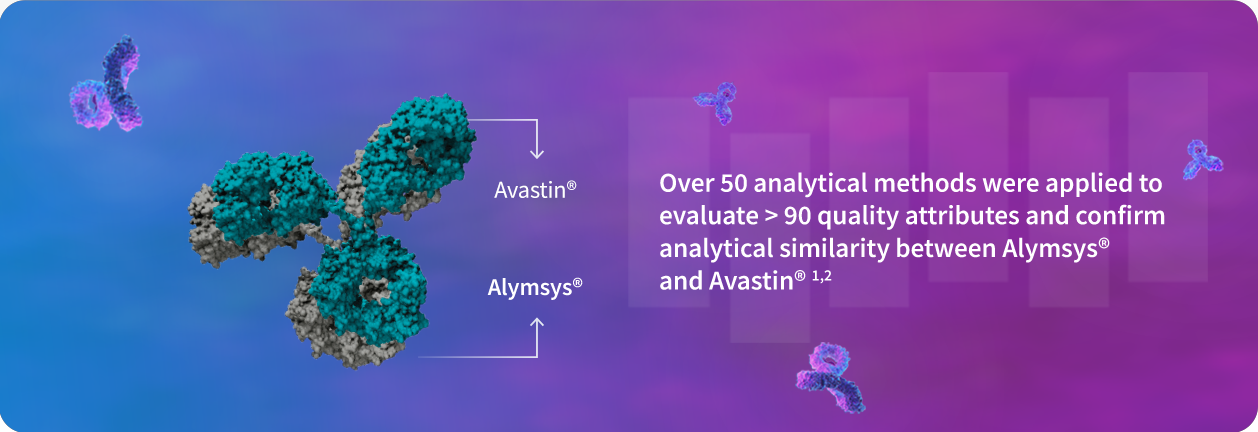
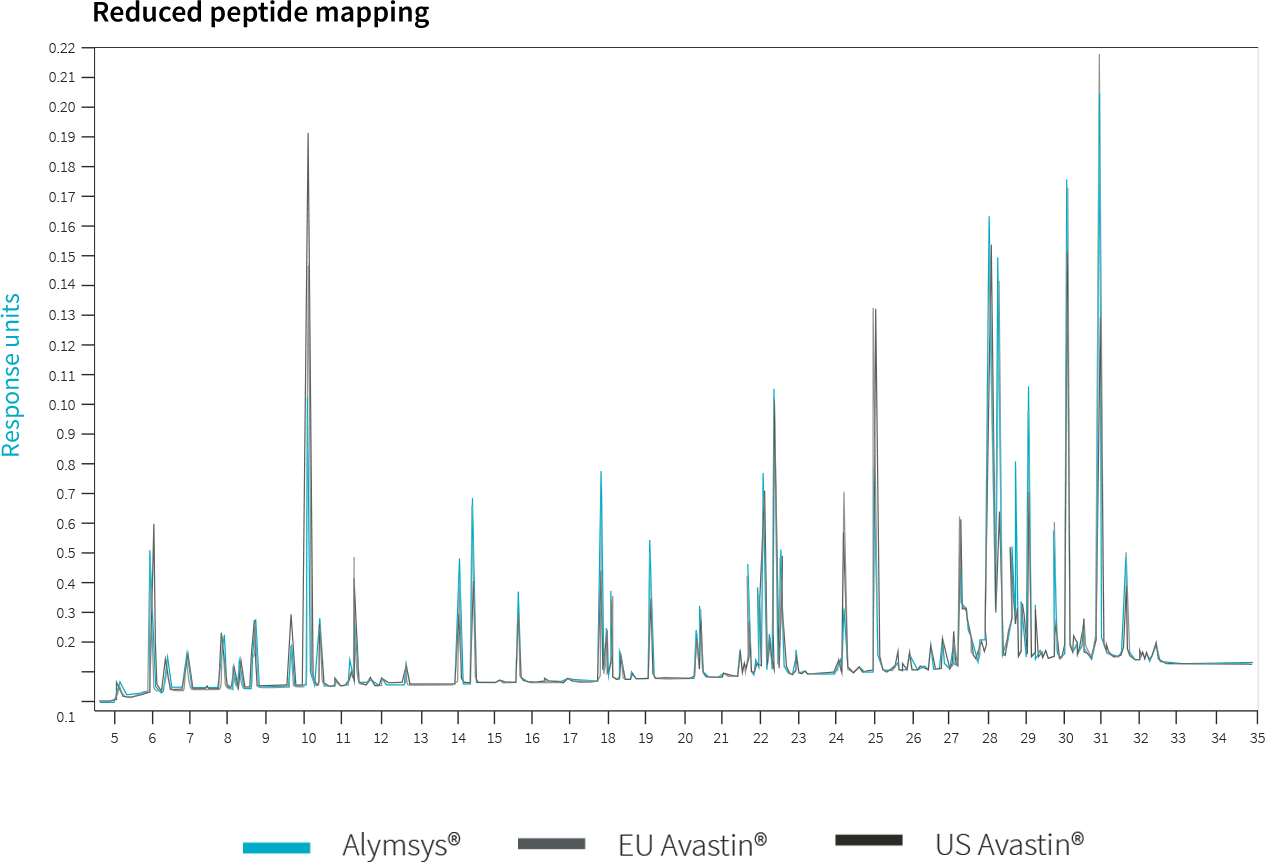
1. Ruppen I, Beydon ME, Solís C, Sacristán D, et al. Similarity demonstrated between isolated charge variants of MB02 , a biosimilar of bevacizumab, and Avastin® following extended physicochemical and functional characterization. Biologicals. 2021;73:41-56. doi:10.1016/j.biologicals.2021.09.001
2. FDA Summary Basis of Approval of MB02 (SBoA). April 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761231Orig1s000TOC.cfm.
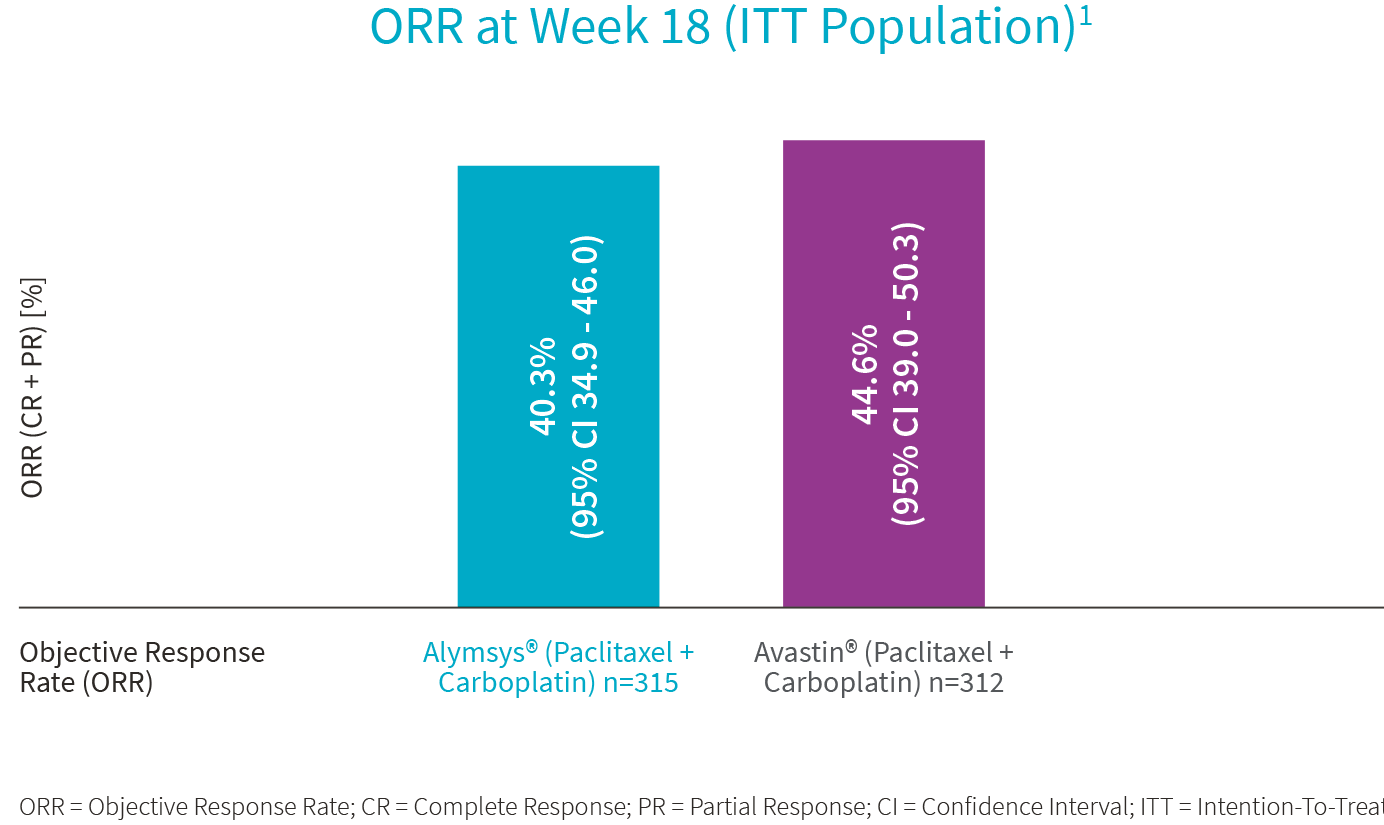
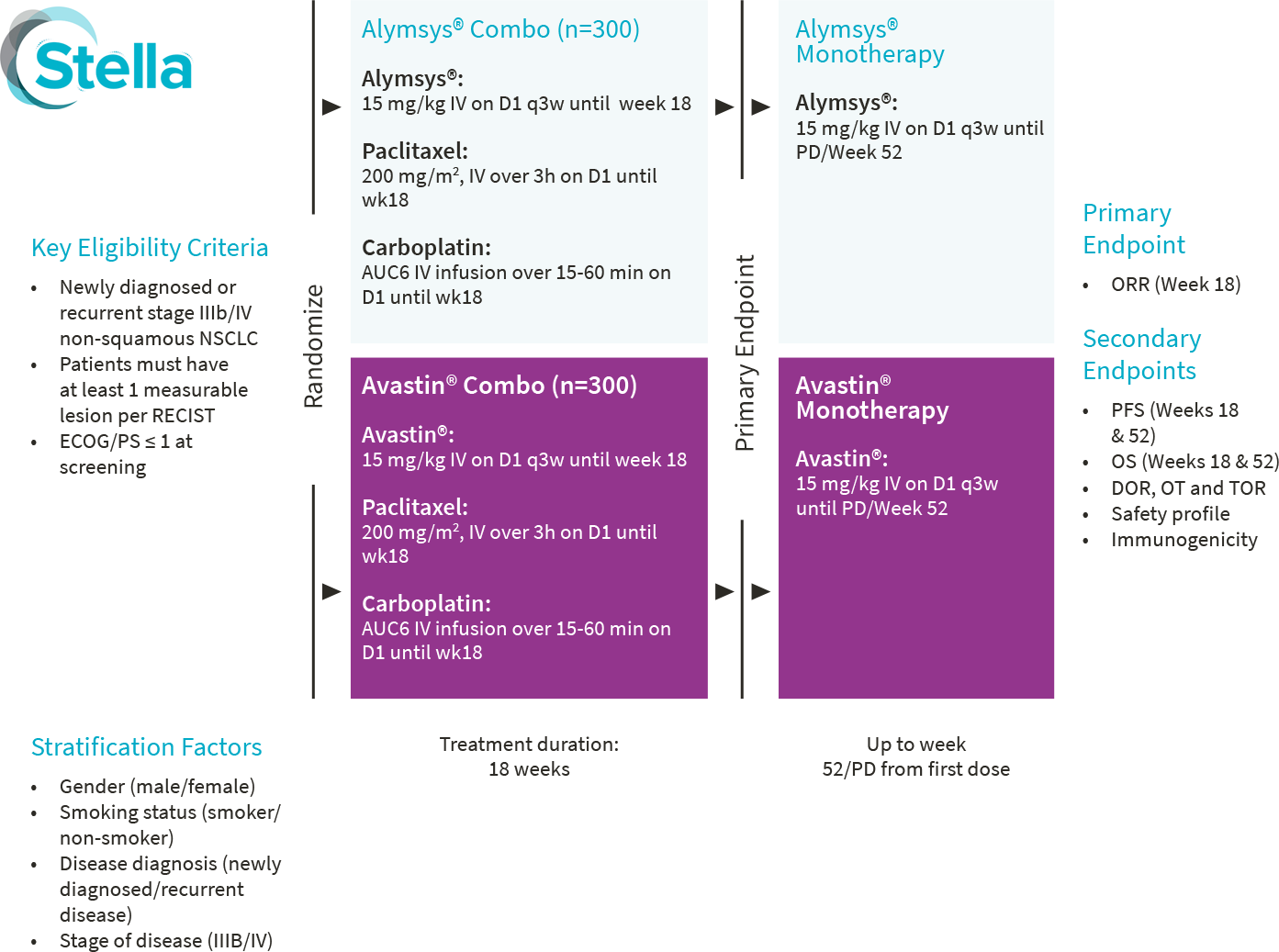
3. Trukhin D, Poddubskaya E, Andric Z, et al. Efficacy, Safety and Immunogenicity of MB02 (Bevacizumab Biosimilar) versus Reference Bevacizumab in Advanced Non-Small Cell Lung Cancer: A Randomized, Double-Blind, Phase III Study (STELLA). BioDrugs. 2021;35(4):429-444. doi:10.1007/s40259-021-00483-w
4. Sinn, A, García-Alvarado, F, Gonzalez, V, Huerga, C, Bullo, F. A randomized, double blind, single dose, comparative study of the pharmacokinetics, safety and immunogenicity of MB02 (bevacizumab biosimilar) and reference bevacizumab in healthy male volunteers. Br J Clin Pharmacol. 2022;88(3):1063- 1073. doi:10.1111/bcp.15032
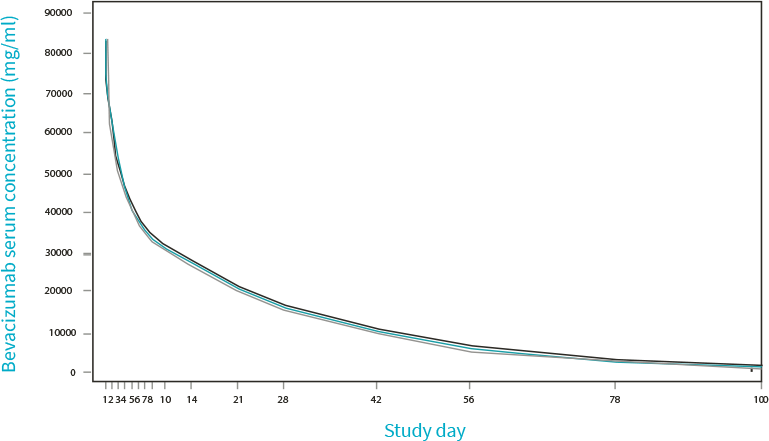
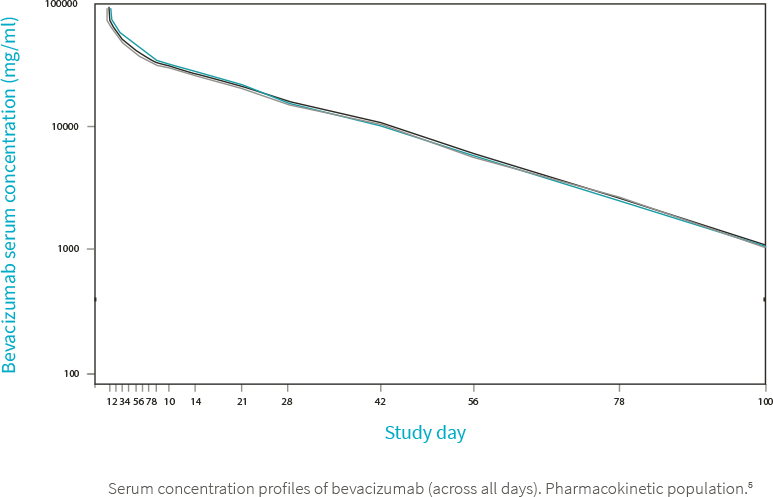
5. Eto T, Karasuyama Y, González V, Del Campo García A. A randomized, single-dose, pharmacokinetic equivalence study comparing MB02 (proposed biosimilar) and reference bevacizumab in healthy Japanese male volunteers. Cancer Chemother Pharmacol. 2021;88(4):713-722. doi:10.1007/s00280-021-04324-z
6. Romera, A. et al. Bevacizumab biosimilar BEVZ92 versus reference bevacizumab in combination with FOLFOX or FOLFIRI as first-line treatment for metastatic colorectal cancer: a multicentre, open-label, randomised controlled trial. Lancet Gastroenterol Hepatol. 2018;3(12):845-855. doi:10.1016/S2468-1253(18)30269-3
7. ALYMSYS. Prescribing information. Valorum Biologics Pharmaceuticals LLC; 2026.